
Sidebar

madamm
Table of Contents
MADAMM
Operating systems: MacOS and Linux.
Contacts : Nuno Sousa Cerqueira Pedro A. Fernandes Maria João Ramos
Citation :
MADAMM: a multistaged docking with an automated molecular modeling protocol.
N. M. F. S. A. Cerqueira, N. F. Bras, P. A. Fernandes AND M. J. Ramos
Proteins (Structure, Function, Bioinformatics), 2009, 74(1):192–206
DOI:10.1002/prot.22146
1. Introduction
Computational capacity has increased dramatically over the last decade making possible the use of more sophisticated and computationally intensive methods in computer-assisted drug design. However, dealing with receptor flexibility in docking methodologies is still a thorny issue. The main reason behind this difficulty is the large number of degrees of freedom that have to be considered in this kind of calculations. However, neglecting it, leads to poor docking results in terms of binding pose prediction in real-world settings.
In order to overcome these limitations we present an automated procedure called MADAMM that allows flexibilization of both the receptor and the ligand during a Multi stAged Docking with an Automated Molecular Modeling protocol. Generally speaking the software uses standard docking software and molecular mechanics force fields in the core process and a set of scripts that automates the process without the intervention of the user. In order to simplify the use of MADAMM a graphical interface has also been developed.
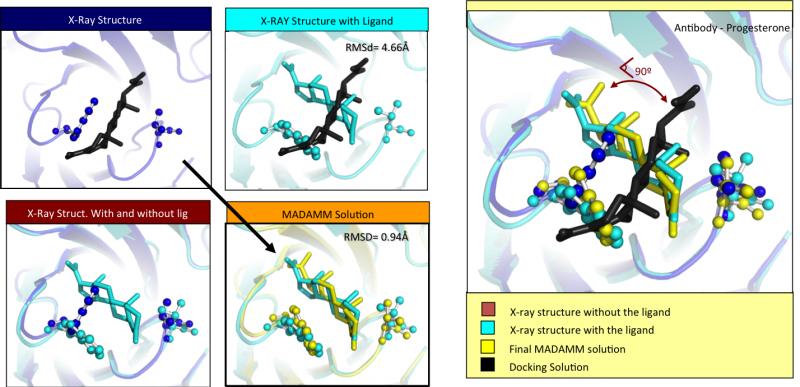
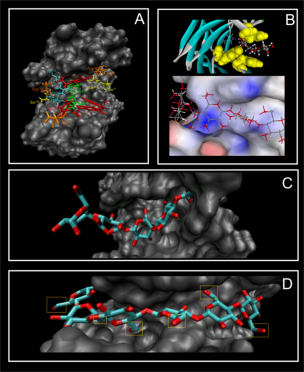
The results obtained with this methodology show that this protocol can lead to dramatic improvements in both sampling and scoring over conventional single rigid protein docking. We observe that the orientation of particular residues, at the interface between the protein and the ligand, have a crucial influence on the way they interact.
At the moment the results indicate that MADAMM can be viewed as a powerful tool for investigating ligand binding poses, allowing the researcher to understand the importance of protein flexibility during the binding processes of the ligands. Moreover this program can be viewed as a valuable tool to predict the binding of ligands in receptors where no experimental data is available.
2. ScreenShots
 |  | 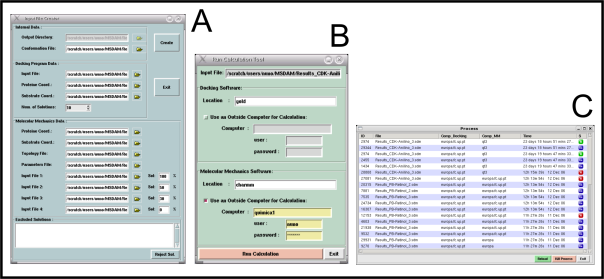 | 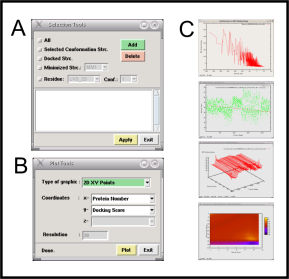 |
3. Download
In order to run the vsÇab plug-in, the Visual Molecular dynamics software (VMD) must be installed. It can be found at http://www.ks.uiuc.edu/Research/vmd/ .
- MADAMM : v1.0
4. Installation
MADAMM can only be installed in LINUX and MAC operating systems, due to the limitations of the GOLD and Charmm software.
To install MADAMM just download the zip file and extract it using the following command:
tar -xvzf MADAMM.tar.gz
After the extraction go to the directory MADAMM and run the file:
cd MADAMM ./MADAMM.sh
4.1. System Dependencies
MADAMM supports computers running MacOS-X, Unix, or Windows, and is distributed free of charge. However at the moment it is advisable to run MADAMM in LINUX systems. In order to use MADAMM you need to install in your computer the following software:
- tcl/tk
In most of the LINUX distributions tcl/tk is already installed in the system. You can check it writing tclsh in your terminal. Even so, it is recommended the installation of the activestate tcl/tk that already comes with the modules that are necessary to run MADAMM. You can download it from http://www.activestate.com/ (Just follow the direction : download>language distributions>activetcl).
To install active state you just have to write in the terminal:
tar -xvzf ActiveTcl8.4.14.0.272572-linux-ix86.tar.gz cd ActiveTcl8.4.14.0.272572-linux-ix86/ ./install.sh (note : you must have root permissions for this command)
- VMD
MADAMM has a small interface that allows to analyse all the results using VMD. VMD is a molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting. You can get VMD from http://www.ks.uiuc.edu/Research/vmd/.
- gnuPLOT
MADAMM has a small interface that allows to plot severall results in different formats using gnuPlot. gnuPlot is a portable command-line driven interactive data and function plotting utility.You can get gnuPlot from: http://www.gnuplot.info/
- GOLD and CHARMm
You need these proprietary software to run the docking software and the molecular mechanics forcefield that are the required by MADAMM.
At the moment the program can only use these two programs but in future releases it will be possible to use also AutoDock and GROMACS and AMBER as molecular mechanic force fields.
madamm.txt · Last modified: 2013/10/03 09:56 by nuno
Except where otherwise noted, content on this wiki is licensed under the following license: CC Attribution-Share Alike 3.0 Unported
