A Química Computacional - Grupo de Bioquímica Computacional
Faculdade de Ciências, Universidade do Porto
NOVAS METODOLOGIAS
No
Grupo de Bioquímica Computacional temos vindo a
desenvolver novos métodos para efectuar simulações moleculares. Esses
métodos são depois livremente disponibilizados à comunidade científica,
para que qualquer pessoa do meio académico as possa utilizar.
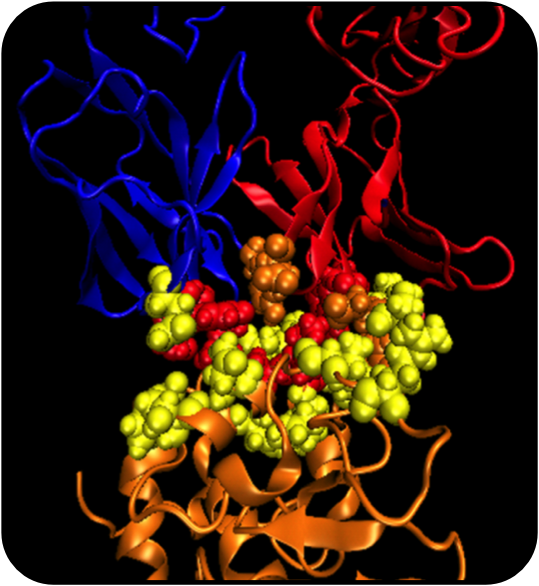
Desenvolvemos um método para estudar a associação entre proteínas. O método calcula qual é a importância, em termos de energia, de cada aminoácido na interface de contacto entre duas proteínas. Os fármacos são desenhados para interactuar com as regiões mais importantes.
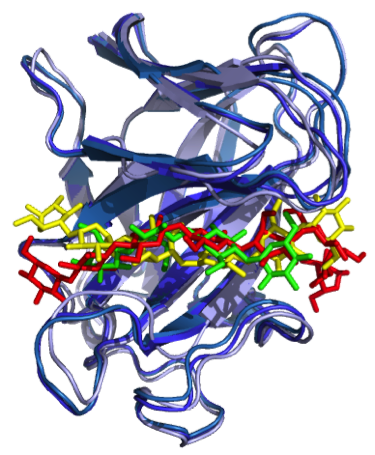
Um outro método que desenvolvemos visa prever a geometria de associação entre um receptor (uma grande molécula) e um ligando (uma pequena molécula). Este processo denomina-se docking. O nosso algoritmo consegue tomar em consideração quer a flexibilidade intrínseca do receptor quer a fleibilidade intrínseca do ligando.
No Grupo de Bioquímica Computacional temos um resumo das metodologias desenvolvidas.
Para mais informações sobre os nossos estudos a este nível consulte o portal do grupo.
Desenvolvemos um método para estudar a associação entre proteínas. O método calcula qual é a importância, em termos de energia, de cada aminoácido na interface de contacto entre duas proteínas. Os fármacos são desenhados para interactuar com as regiões mais importantes.
Um outro método que desenvolvemos visa prever a geometria de associação entre um receptor (uma grande molécula) e um ligando (uma pequena molécula). Este processo denomina-se docking. O nosso algoritmo consegue tomar em consideração quer a flexibilidade intrínseca do receptor quer a fleibilidade intrínseca do ligando.
{kind=link}
Mutagénese
de Varrimento por Alaninas, um método para explorar a importância de
cada aminoácido na associação de duas proteínas. A cor indica a
importância do aminoácido, do amarelo (menos importante) para o
vermelho (mais importante).
Clique sobre a imagem para visualizar uma animação que simula a abertura de um complexo entre proteínas, para melhor visualização da sua interface.
Clique sobre a imagem para visualizar uma animação que simula a abertura de um complexo entre proteínas, para melhor visualização da sua interface.
Software MADAMM, um poderoso algoritmo de docking molecular que prevê a geometria de associação entre um receptor e um ligando.
Clique sobre a imagem para ver uma animação do processo de docking.
Clique sobre a imagem para ver uma animação do processo de docking.
No Grupo de Bioquímica Computacional temos um resumo das metodologias desenvolvidas.
Para mais informações sobre os nossos estudos a este nível consulte o portal do grupo.